41. Wissenschaftliche Jahrestagung der Deutschen Gesellschaft für Phoniatrie und Pädaudiologie (DGPP)
Relevanz der genetischen Diagnostik bei kindlichen Hörstörungen
Abstract
Hintergrund: Neben der pädaudiologischen Diagnostik als Grundlage zur frühzeitigen optimalen Versorgung ist die Ursachenklärung einer Hörstörung im Kindesalter für den Behandlungserfolg relevant, teilweise auch therapieentscheidend, jedoch noch nicht klinischer Standard. Die Möglichkeit durch eine humangenetische Abklärung früh Komorbiditäten zu erkennen und ggf. präventive Maßnahmen zu ergreifen, ist gerade bei syndromalen Hörstörungen anzustreben.
Material und Methoden: In einer retrospektiven Analyse wurden genetische Befunde von Januar 2016 bis Dezember 2024 von mit Hörstörungen assoziierten Genen als Stufendiagnostik mit Stufe 1 = GJB2/GJB6; Stufe 2 = MTRNR1/MTTL1; Stufe 3 = Next Generation Sequencing (NGS) untersucht. Das Alter bei Erstdiagnose, der Schweregrad der Hörstörung, der technischen Versorgung und Benefit mittels altersnormierten Tests zum Sprachverstehen, Sprachverständnis, IQ-Test sowie mögliche Auffälligkeiten in der Bildgebung (CT/MRT) wurden unter genetischen Aspekten analysiert.
Ergebnisse: Insgesamt erfolgte bei 287 schwerhörigen Kindern (52% Jungen; 48% Mädchen) eine Blutentnahme zur humangenetischen Untersuchung, wovon 41,81% (n=120) einen genetisch auffälligen Befund aufwiesen. Das Alter bei Erstdiagnosestellung lag zwischen 3 Monaten und 18 Jahren. In Stufe 1 (n=256) waren 20,7% auffällig (n= 53). Von den in Stufe 1 unauffälligen Befunden (n=203) wurde bei 66,5% (n=135) eine ergänzende Testung vorgenommen und bei 68 Kindern die Interpretation der humangenetischen Diagnostik mit der unauffälligen Stufe 1 belassen. In Stufe 2 waren von 167 getesteten Kindern 46,7% auffällig (n=78 vs. 86 unauffällige Befunde). Da bei 11 Kindern sowohl Stufe 1 und 3 auffällig waren, ergab sich die Summe auffälliger genetischer Befunde auf 131 bei 120 untersuchten Kindern. Von 120 auffälligen Befunden waren 71,6% (n=86) mit einer Pathogenitätsstufe 4 oder 5 und damit ursächlich für die Hörstörung. Es wurden 46 verschiedene Gene gefunden, davon ursächlich für syndromale Hörstörungen z.B. STRC (n=12, auch in Kombination mit CATSPER2 für das Taubheits-Infertilitäts-Syndrom), MYO7A für Usher-Syndrom (n=7), WFS1 für Wolfram-Syndrom (n=5), GATA3 für Barakat-Syndrom (n=3).
Schlussfolgerungen: Bei unserem Studien-Kollektiv konnte die Hörstörung bei 41,81% einer genetischen Ursache zugeordnet werden. Bei 43,7% (n=59) der in Stufe 1 unauffälligen Kinder ergab eine ergänzende genetische Untersuchung bis Stufe 3 (bei aktuell 326 zu untersuchende Gene im NGS-Panel) doch noch einen auffälligen Befund.
Text
Hintergrund
Neben Neugeborenen-Infektionen wie CMV belaufen sich genetische Ursachen für angeborene Hörstörungen auf bis zu 50% [1]. Davon sind ungefähr 70% nicht-syndromaler und 30% syndromaler Genese [2]. Neben der pädaudiologischen Diagnostik als Grundlage zur frühzeitigen optimalen Versorgung ist die Ursachenklärung einer Hörstörung im Kindesalter für den Behandlungserfolg relevant, teilweise auch therapieentscheidend, jedoch noch nicht klinischer Standard. Die Möglichkeit durch eine humangenetische Abklärung früh Komorbiditäten zu erkennen und ggf. präventive Maßnahmen zu ergreifen, ist gerade bei syndromalen Hörstörungen anzustreben.
Material und Methoden
In einer retrospektiven Analyse wurden bei Kindern mit einer gesicherten organischen Hörstörung von Januar 2012 bis Dezember 2024 genetische Befunde von mit Hörstörungen assoziierten Genen untersucht. Dabei folgt die genetische Untersuchung einer Stufendiagnostik: In Stufe 1 werden die Gene GJB2 sowie GJB6 mittels PCR untersucht. Die Stufe 2 untersucht die mitochondrialen Gene MTRNR1 und MTTL. Mittels Next Generation Sequencing (NGS) werden seit Anfang der 2010er Jahre durch die Stufe 3 immer neue Gene komplettiert.
Das Alter bei Erstdiagnose, der Schweregrad der Hörstörung, die technische Versorgung und deren Benefit mittels altersnormierter Tests zum Sprachverstehen, Sprachverständnis, IQ-Test sowie mögliche Auffälligkeiten in der Bildgebung (CT-Felsenbein/cMRT) wurden unter genetischen Aspekten analysiert.
Ergebnisse
Insgesamt erfolgte bei 540 schwerhörigen Kindern (52% Jungen und 48% Mädchen) eine Blutentnahme zur humangenetischen Untersuchung, wovon 43,33% (n=234) einen genetisch auffälligen Befund aufwiesen. Das Alter bei Erstdiagnosestellung lag zwischen 3 Monaten und 18 Jahren.
In Stufe 1 waren von 415 getesteten Kindern 24,3% auffällig (n=101). Von den in Stufe 1 unauffälligen Befunden (n=314) wurde bei 72,3% (n=227) eine ergänzende Testung vorgenommen und bei 87 Kindern die Interpretation der humangenetischen Diagnostik mit der unauffälligen Stufe 1 belassen. In Stufe 2 waren von 118 getesteten Kinder nur 3 Kinder auffällig. In Stufe 3 waren von 278 getesteten Kindern 50% auffällig (n=139). Bei 12 Kindern ergab sich sowohl in Stufe 1 als auch in Stufe 3 ein auffälliger Befund. Somit ergab sich die Summe von 246 auffälligen genetischen Befunden bei 234 auffälligen Kindern (siehe Abbildung 1 [Abb. 1]).
Abbildung 1: Prozentuale Verteilung der Ergebnisse der genetischen Befunde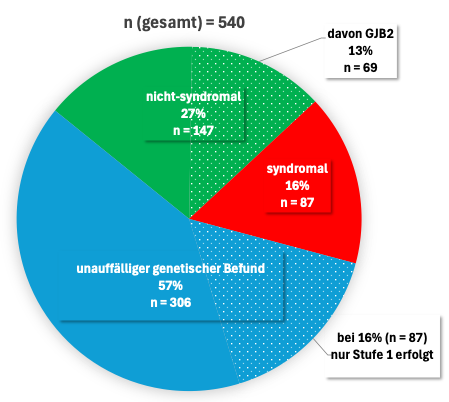
Eine einseitige Hörstörung lag bei 20,2% (n=109) der Kinder vor (einseitig rechts: 52 und einseitig links: 57). Dabei lag nur bei 8 Kindern ein auffälliger genetischer Befund mit einer Pathogenitätsstufe von maximal 3 und damit nicht sicher krankheitsverursachend vor. Insgesamt lag bei 60 Kindern eine einseitige Taubheit (SSD) vor. Bildmorphologisch wurde diese mittels cMRT bei insgesamt 56 Kindern untersucht. Dabei ergab sich bei 45% (n=27) eine auffällige Bildgebung mit entweder hypoplastischen oder nicht darstellbaren ipsilateralen N. cochlearis.
Eine beidseitige Surditas lag bei 85 Kindern vor. Dabei lag bei 58% (n=50) ein auffälliger genetischer Befund vor.
Insgesamt waren 265 Kinder beidseits mit Hörgeräten und 61 Kinder beidseits mit Cochlea-Implantaten versorgt. 36 Kinder waren bimodal versorgt und 51 Kinder waren beidseits unversorgt.
Von 234 auffälligen Befunden waren 51,28% (n=120) mit einer Pathogenitätsstufe 4 oder 5 und damit ursächlich für die Hörstörung. Es wurden 46 verschiedene Gene gefunden. Davon waren 20 Gene ursächlich für syndromale Hörstörungen. In absteigender Häufigkeit ergaben sich folgende Syndrome mit deren auslösenden Genen: Usher-Syndrom (n=25, davon 10x MYO7A; 5x ADGRV1; 5x USH2A; 4x CDH23; 1x USH1G), Taubheits-Infertilitäts-Syndrom (n=19, STRC, auch in Kombination mit CATSPER2), Wolfram-Syndrom (n=7, WFS1), Waardenburg-Syndrom (n=7, davon 6x MITF, 1x PAX3), Pendred-Syndrom (n=5, SLC26A4), Branchio-oto-renales (BOR)-Syndrom (n=5, davon 4x SIX1, 1x EYA1), Stickler-Syndrom (n=5, davon 4x COL11A2 ,1x COL11A1), Barakat-Syndrom (n=4, GATA3), CHARGE-Syndrom (n=4, CHD7), Alport-Syndrom (n=3, 2x COL4A3, 1x COL4A5), Jervell-Lange-Nielson-Syndrom (n=1, KCNQ1), Treacher-Collin-Syndrom (n=1, TCOF1), Noonan-Syndrom (n=1, PTPN11). Insgesamt zeigten sich bei 37,17% (n=87) der Kinder eine syndromale Genese der Hörstörung (siehe Abbildung 2 [Abb. 2]).
Abbildung 2: Alphabetische Auflistung mit absoluten Zahlen der genetisch gesicherten syndromalen Hörstörungen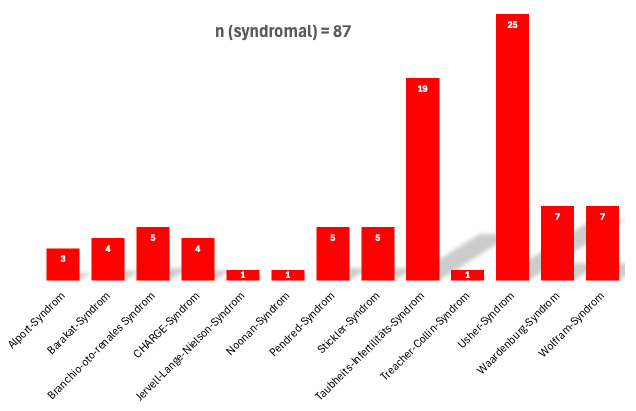
Schlussfolgerungen
Bei unserem Studien-Kollektiv konnte die Hörstörung bei 43% (n=234) einer genetischen Ursache zugeordnet werden. Bei 110 der in Stufe 1 unauffälligen Kinder ergab eine ergänzende genetische Untersuchung bis Stufe 3 (bei aktuell 326 zu untersuchende Gene im NGS-Panel) doch noch einen auffälligen Befund.
Wenn von den 87 nicht ergänzend untersuchten Kindern knapp die Hälfte in Stufe 3 auffällig wären, könnte somit angenommen werden, dass die Anzahl der genetisch auffälligen Kinder insgesamt von 43% auf rund 51% steigen würde.
Die in dieser Studie erhobenen Daten von Kindern mit genetisch gesicherter Hörstörungen bilden eine Ergänzung zu den in der Literatur bisher erhobenen Daten, da dort vor allem die Performance selektierter Erwachsener mit Hörstörungen nach Cochlea-Implantat beschrieben wird [3].
Dies kann die Beratung der Eltern von schwerhörigen Kindern zur optimalen Versorgung vereinfachen und bei frühzeitiger Ursachenklärung auch dabei helfen, präventive Maßnahmen bei möglichen Komorbiditäten zu ergreifen.
References
[1] Young A, Ng M. Genetic Hearing Loss. In: StatPearls. Treasure Island (FL): StatPearls; 2025. Verfügbar unter: https://www.ncbi.nlm.nih.gov/books/NBK580517/[2] Kubisch C. Genetische Grundlagen nichtsyndromaler Hörstörungen. Dtsch Arztebl. 2005;102(43):A 2946–2952.
[3] Tropitzsch A, Schade-Mann T, Gamerdinger P, Dofek S, Schulte B, Schulze M, Fehr S, Biskup S, Haack TB, Stobe P, et al. Variability in Cochlear Implantation Outcomes in a Large German Cohort With a Genetic Etiology of Hearing Loss. Ear Hear. 2023;44(6):1464-84.

