41. Wissenschaftliche Jahrestagung der Deutschen Gesellschaft für Phoniatrie und Pädaudiologie (DGPP)
Numerische Chromosomenaberrationen als Ursache von Sprachentwicklungsstörungen
Abstract
Hintergrund: Numerischen Chromosomenaberrationen, auch Genommutation genannt, sind oft unerkannte Ursachen von Sprachentwicklungsstörungen oder ausbleibenden Spracherwerb. Die häufigste Chromosomenaberrationen ist die Trisomie 21, welche weltweit bei 1:800 Geburten auftritt. Weniger bekannt sind das Klinefelter-Syndrom mit einem Chromosomensatz von 47, XXY und einer Prävalenz von 1:500 bis 1:1000 männlichen Lebendgeburten oder Tetrasomie X, 48, XXXX mit Prävalenz von <1/1.000.000.
Material und Methoden: Case Report eines 6-jährigen Jungen, der bis dato bei unbekannter Ursache einer schweren Entwicklungsstörung mit mentaler Retardierung, ausbleibendem Spracherwerb zur weiteren phoniatrischen Mitbehandlung in unserem phoniatrisch-pädaudiologischen SPZ vorgestellt wurde. Neben einer pädaudiologischen Diagnostik inkl. BERA-Messung wurde durch uns eine Chromosomenanalyse und humangenetische Diagnostik auf Entwicklungsstörungen bzw. Intelligenzpanel beauftragt.
Ergebnisse: Mittels der Chromosomenanalyse konnte innerhalb von 3 Wochen die Ursache der schwerwiegenden Mehrfachbehinderung des Jungen mit ausstehenden Spracherwerb nach Jahren geklärt werden. Es zeigte sich eine Chromosomensatz von 49, XXXXY (ICD 10: Q98.1), welches mit einer Prävalenz von neugeborenen Knaben zwischen 1:85.000 und 1:100.000 angegeben wird. Die betroffenen Kinder fallen typischerweise durch faciale Dysmorphien (Hypertelorismus, große flache Nase mit niedrigem Nasenrücken, mongoloide Lidspalten, Epikanthus, Progenie, gefaltete Ohren), einem fortschreitenden mittelschweren bis schweren geistigen Abbau mit zunehmendem Alter (IQ 20–70), kurzer Hals, Cubitus valgus, Pes planus, Klinodaktylie der Kleinfinger und überstreckbare Gelenke und durch fehlenden Hochwuchs auf. Durch die genetische Beratung konnte die präventive Mitbehandlung durch Kinderkardiologen, -nephrologen, -orthopäden, Augenärzte eingeleitet werden. Für die betroffene Familie konnte so nicht nur die Ursache geklärt, sondern eine Perspektive zur Weiterbehandlung und Selbsthilfe sowie versorgungsmedizinische Unterstützung geschaffen werden.
Schlussfolgerungen: Eine molekulargenetische Analyse mittels Array-CGH kann bei ausstehendem Spracherwerb mit unklarer mentale Retardierung (IQ<70) und mehrere dysmorphologische Merkmale und/der Verdacht auf Autismus sowie multiple angeborene Fehlbildungen unklarer Ätiologie zur Abklärung numerischer Chromosomenaberrationen genutzt werden. Die Behandlung der Patienten sollte durch ein multidisziplinäres Expertenteam inkl. Phoniater eines ZSE erfolgen.
Text
Hintergrund
Numerische Chromosomenaberrationen, auch Genommutation genannt, sind oft unerkannte Ursachen von Sprachentwicklungsstörungen oder ausbleibenden Spracherwerb. Die häufigste Chromosomenaberrationen ist die Trisomie 21, welche weltweit bei 1:800 Geburten auftritt. Weniger bekannt sind das Klinefelter-Syndrom mit einem Chromosomensatz von 47, XXY (oder als Mosaikform 46, XY/47, XXY/6%) und einer Prävalenz von 1:500 bis 1:1.000 männlichen Lebendgeburten [1], [2] oder Tetrasomie X, 48, XXXX mit einer Prävalenz von <1/1.000.000.
Das mütterliche Alter ist der einzige evidenzbasierte Risikofaktor für das Klinefelter-Syndrom: Ab einem mütterlichen Alter von 40 Jahren ist die Wahrscheinlichkeit, ein Kind mit Klinefelter-Syndrom zu bekommen, viermal so hoch wie bei Frauen unter dem 24. Lebensjahr. Seit September 2020 wurden von der European Academy of Andrology erstmals eine Leitlinie für das Klinefelter-Syndrom veröffentlicht [3].
Hochgradigere X-Chromosomenaneuploidien z.B. 48, XXXY, 48, XXYY oder 49,XXYYY-Polysomien werden oft als Varianten des Klinefelter-Syndroms bezeichnet, jedoch ist ihr Auftreten weitaus seltener (1:18.000–1:100.000 der männlichen Geburten) [4]. Diese Varianten verursachen ein im Vergleich zur klassischen 47,XXY-Form schwerwiegenderes Erkrankungsbild [2].
Material und Methoden
Case Report eines aus Afghanistan stammenden 6-jährigen Jungen, der bis dato bei unbekannter Ursache einer schweren Entwicklungsstörung mit mentaler Retardierung, nahezu ausbleibendem Spracherwerb ohne ambulante Förderung bei anstehender Beschulungspflicht zur weiteren phoniatrischen Mitbehandlung in unserem phoniatrisch-pädaudiologischen SPZ vorgestellt wurde. Neben einer pädaudiologischen Diagnostik inkl. BERA-Messung wurde durch uns eine Chromosomenanalyse und humangenetische Diagnostik auf Entwicklungsstörungen bzw. Intelligenzpanel beauftragt.
Ergebnisse
Das 6. Kind von 8 Kindern der Familie sei im Heimatland mit zu leichtem Geburtsgewicht spontan entbunden worden und erhielt postnatal eine Antibiose. Nach Migration in die BRD im 10/2022 erfolgte zur weiteren Behandlung eine Vorstellung in einem anderen SPZ sowie unauffällige EEG-Messung bei Mikrocephalie. Eine weit unterdurchschnittliche Intelligenz wurde im SON-R 2-8 bestätigt.
Die Geschwister seien gesund, die Eltern nicht konsanguin.
Subjektive audiologische Untersuchungen waren im ambulanten Setting nur unzureichend umsetzbar, so dass keine Aussage zum Hören getroffen werden konnte (Reaktionen im Freifeld bei 45–65 dB). Der Nachweis von TEOAE bds. Gelang bei Tubenbelüftungsstörung nicht. Im stationären Setting erfolgte eine BERA-Messung mit Click-Schwellen bei 45 dB bds. Sowie bei 1/4 kHz mittels Toneburst AEP Welle JV bei rechts 40/70 dB und links 30/60 dB sowie tonaudiometrisch mit eigenen Angaben nach mehrtägigem Üben mit Hochtonsteilabfall (Abbildung 1 [Abb. 1]), so dass eine vergleichende Hörgeräteanpassung bds. erfolgte. Hierbei zeigte sich ein sehr schöner Hörgewinn.
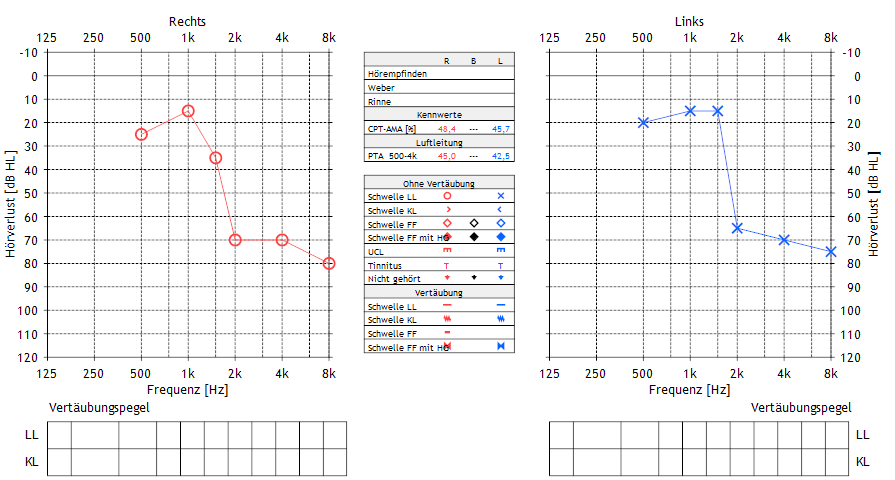
Zudem fiel eine juvenile Dysphonie, faciale Dysmorphie mit tief angesetzten Ohrmuscheln, breiter Nasenwurzel, weitem Augenabstand und Vierfingerfurche rechts, Karies und Non Descensus testis bds. sowie stimulierende Geräusche auf.
In der logopädischen Diagnostik ergaben sich folgende Befunde:
- TROG-D – Versuch auf Deutsch und Pashtu: Deutsch wird gar nicht verstanden. Das Kind reagierte nur bei Pashtu – übersetzt durch den Vater: Abbruch nach 5 Blöcken: orientierend RW = 0; PR = k.A. Es kam im Verlauf zu einer Selbstkorrektur.
- Lautbefund: nicht durchführbar mangels Mitarbeit/Wortschatz: die Aufgabenstellung wird in beiden Sprachen nicht verstanden. Er wiederholt von sich aus auch keine Wörter, sagt nur „la“, wenn die Therapeutin auf etwas zeigte oder vollkommen Unverständliches, dass auch der Vater nicht verstand. Auch bei einer gemeinsamen Bilderbuchbetrachtung war keine verständliche spontansprachliche Äußerung zu beobachten. Die beobachteten expressiven Äußerungen auf Pashtu seien auch lautlich verändert: V.a. höhergradige Störung im Bereich Phonetik-Phonologie.
- Grammatik: max. 2-Wortäußerungen (Nomen + Nomen oder Nomen + Verb) laut Vater. Grammatikalische Fähigkeiten sind daher noch nicht eruierbar: Schwerstgradige Störung der syntaktisch-morphologischen Fähigkeiten.
- Wortschatz: max. 30 Wörter auf Pashtu laut Vater u.a. alle Namen der Familienmitglieder (ca. 10) und einige Verwandtschaftsbezeichnungen (Oma, Onkel, Papa, Mama) und andere Wörter, die spontan angegeben werden können: „Wasser“, „komm“.
Mittels Chromosomenanalyse (aus Lymphozytenkulturen) konnte innerhalb von 3 Wochen (in 31 ausgewerteten Metaphasen) die Ursache der schwerwiegenden Mehrfachbehinderung des Jungen mit nahezu ausstehenden Spracherwerb nach Jahren geklärt werden: Es zeigte sich ein Chromosomensatz von 49, mit den Gonosomen XXXXY (ICD 10: Q98.1) – also drei überzähligen X. Dies wird mit einer Prävalenz von neugeborenen Knaben zwischen 1:85.000 und 1:100.000 angegeben und durch eine maternale Nondisjunction in der Keimzellentwicklung verursacht.
Die betroffenen Kinder fallen typischerweise durch faciale Dysmorphien (Hypertelorismus, große flache Nase mit niedrigem Nasenrücken, mongoloide Lidspalten, Epikanthus, Progenie, gefaltete Ohren), einem fortschreitenden mittelschweren bis schweren geistigen Abbau mit zunehmendem Alter (IQ 20–70) und einer erheblich verzögerten Sprachentwicklung, kurzer Hals, Cubitus valgus, Pes planus, Klinodaktylie der Kleinfinger und überstreckbare Gelenke und durch fehlenden Hochwuchs bei Mangel an Wachstumshormon und schwer ausgeprägtem Hypogonadismus auf. Auch angeborene Herzfehler, Skelettanomalien, Hirnfehlbildungen, Nierenhypoplasie, Strabismus und eine schwere progrediente Myopie sind weitere mögliche Symptome (ORPHA: 96264). Chronische Mittelohrentzündungen werden in der Literatur beschrieben und als ursächlich für die Hörstörung diskutiert.
Durch die Diagnosestellung konnte die Mitbehandlung durch Kinderkardiologen, -nephrologen, -orthopäden und Augenärzte eingeleitet werden. Ab Beginn der Pubertät kann der bestehende Testosteronmangel durch eine entsprechende Hormon-Substitutionstherapie in Form von Spritzen, Pflastern oder Gel und ggf. ein Vitamin D-Mangel mit einer Vitamin-D-Substitution ausgeglichen werden. Für die betroffene Familie konnte so nicht nur die Ursache geklärt, sondern eine Perspektive zur Weiterbehandlung und Selbsthilfe sowie versorgungsmedizinische Unterstützung geschaffen werden.
Schlussfolgerungen
Eine molekulargenetische Analyse mittels Array-CGH kann bei ausstehendem Spracherwerb mit unklarer mentale Retardierung (IQ <70) und mehrere dysmorphologische Merkmale und/der Verdacht auf Autismus sowie multiple angeborene Fehlbildungen unklarer Ätiologie zur Abklärung numerischer Chromosomenaberrationen genutzt werden. Die Behandlung der Patienten sollte durch ein multidisziplinäres Expertenteam inkl. Phoniater eines Zentrums für seltene Erkrankungen (ZSE) erfolgen.
References
[1] Nieschlag E. Klinefelter-Syndrom. Häufigste Form des Hypogonadismus, aber oft übersehen oder unbehandelt. Dtsch Arztebl Int. 2013;110(20):347-53. DOI: 10.3238/arztebl.2013.0347[2] Tüttelmann F, Gromoll J. Novel genetic aspects of Klinefelter's syndrome. Mol Hum Reprod. 2010 Jun;16(6):386-95. DOI: 10.1093/molehr/gaq019
[3] Zitzmann M, Aksglaede L, Corona G, Isidori AM, Juul A, T'Sjoen G, Kliesch S, D'Hauwers K, Toppari J, Słowikowska-Hilczer J, Tüttelmann F, Ferlin A. European academy of andrology guidelines on Klinefelter Syndrome Endorsing Organization: European Society of Endocrinology. Andrology. 2021 Jan;9(1):145-67. DOI: 10.1111/andr.12909
[4] Tartaglia N, Ayari N, Howell S, D'Epagnier C, Zeitler P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011 Jun;100(6):851-60. DOI: 10.1111/j.1651-2227.2011.02235.x
[5] Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab. 2003 Feb;88(2):622-6. DOI: 10.1210/jc.2002-021491

